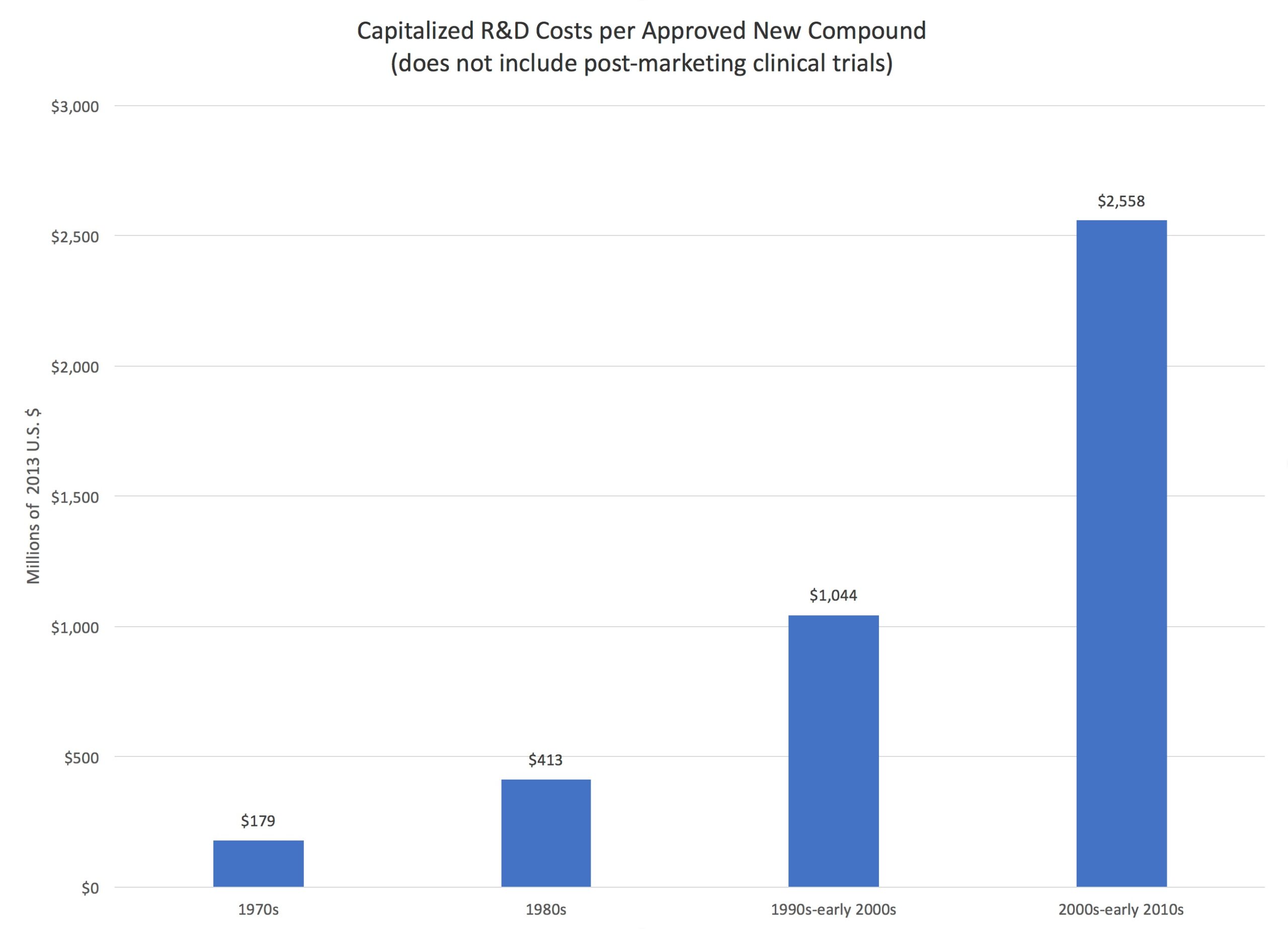
Economists have shown that the cost to get one drug to market successfully is now more than $2.8 billion. This cost has been growing at 7.5 percent per year, more than doubling every ten years. Most of this cost is due to FDA regulation. Some potentially helpful drugs don’t ever make it to market because the cost the company must bear is too high. Drug companies regularly “kill” drugs that could be effective because the potential profits, multiplied by the probability of collecting them, are less than the anticipated costs. One of us has helped kill drugs for brain cancer, ovarian cancer, melanoma, hemophilia and other debilitating conditions. Imagine a drug for melanoma that never got on the market due to FDA regulation. In a sense, its price is infinite because it can’t be purchased. Reduce FDA regulation so that it gets on the market, and the price falls from “infinite” to merely “high.” If you had melanoma, which would you rather have: no drug or a high-priced drug that treats it?
If we simply went back to pre-1962 law, the FDA could still require proof of safety, but would not be able to require evidence on efficacy. This one change would allow drugs to be developed faster–often as much as 10 years faster. Market success would establish efficacy. Could there be ineffective drugs? Sure. But as doctors and patients learn, such drugs would disappear over time. This is nothing new; doctors and patients regularly evaluate drugs for efficacy. Clinical trials often show that perhaps only 20 percent, 40 percent, or 60 percent of patients benefit. Even when the FDA finally approves the drug as “safe and efficacious,” doctors must still evaluate the drug to find out how efficacious it is for each particular patient. In practice, an FDA certification of efficacy is just a starting point.
Who would want to take a drug that has not been shown, to the FDA’s satisfaction, to be effective? Almost everyone. Many drugs have off-label uses. These are uses that doctors have found effective for a particular use but that the FDA has not approved for that use. According to WebMD, “More than one in five outpatient prescriptions written in the U.S. are for off-label uses.” Tabarrok cites studies showing that 80 to 90 percent of pediatric patients are prescribed drugs for off-label uses.
As is well-known in the medical establishment, off-label prescribing is legal and widely practiced. Indeed, Congress, the National Institute for Health, Medicare, the Veterans Administration, and the National Cancer Institute all encourage it. Consider gastroparesis, a poorly understood upper gastrointestinal disorder in which the contents of the stomach do not move efficiently into the small intestine. Diabetics are particularly susceptible to this condition. The FDA has approved only one drug to treat it: metoclopramide. But doctors have found that, for some patients, an antibiotic called erythromycin reduces nausea, vomiting, and abdominal pain. Erythromycin is not FDA-approved to treat gastroparesis. But it works. Moreover, off-label uses in oncology account for as much as 90 percent of all cancer treatments. For some diseases, like AL amyloidosis, there are no approved medicines. Not a single one. So what do doctors do? They use medicines developed to treat related diseases, such as multiple myeloma, even though they and their patients would prefer medicines that treat AL amyloidosis directly.
This is from David R. Henderson and Charles L. Hooper, “Why Are Drug Prices so High?” Goodman Institute Brief Analysis No. 117, January 10, 2017.
READER COMMENTS
Nick Bradley
Jan 14 2017 at 10:31am
Why are you being so deeply dishonest? R&D as a percent of pharma sales has bounced between 15% and 20% for decades; the biggest cost driver is marketing (40%). .
Yet you find a way to justify Sky high prices on one of the most profitable industries
David R. Henderson
Jan 14 2017 at 11:34am
@Nick Bradley,
Why are you being so deeply dishonest?
When did you stop beating your wife?
Mark Cancellieri
Jan 14 2017 at 12:08pm
In my humble, armchair-economist opinion, the three main drivers of high drug prices in no particular order are:
1) Patents
2) FDA regulation
3) The high percentage of third-party payments (i.e. low out-of-pocket spending on drugs).
Scott Sumner
Jan 14 2017 at 12:16pm
One good thing about Trump is that there are rumors he will appoint a deregulation proponent to the FDA.
Nick, Isn’t a lot of that ad money spent trying to convince doctors to use certain drugs? That raises the question of why this spending is effective. Are doctors corrupt, or ignorant? (And I use ‘ignorant’ in the non-pejorative sense of not well-informed on new discoveries since they were in medical school.)
And I agree with Mark’s comment.
Jim Glass
Jan 14 2017 at 12:45pm
Why are you being so deeply dishonest?
the biggest cost driver is marketing (40%)
Marketing isn’t a cost driver *at all*. It increases net revenue. Spend $x on marketing, receive $X+ in revenue. As such, it provides funds *for* product development and, if anything, provides leeway to reduce drug prices (through price discrimination).
Start a business. Don’t market it. Only your mother and three best buddies are willing to buy your product – but not at anywhere near the price you need to cover your startup costs. You are busted. OR instead market it, sell to 10,000 customers at a modest price, reinvest your profits in developing your next products…
So your “cost driver” there going up enables you to slash your prices and fund new product development.
In short, your the quality of your business analysis is on a par with that of your manners.
George
Jan 14 2017 at 1:23pm
@David Henderson
That seems like an overly harsh comment. You are usually polite in your discourse. Did I miss something?
Also you didn’t even acknowledge the comment which if true would seem to suggest that there’s more to the story than the FDA regulation unless of the FDA also regulates marketing
AntiSchiff
Jan 14 2017 at 1:45pm
Dr. Henderson,
I’d prefer to see the FDA abolished. I think the FTC should just enforce laws against fraudulent marketing.
I think total drug legalization, only requiring prescriptions for drugs like antibiotics, for example, could also bring down drug prices. Then, drugs bottles which currently get filled and checked by no fewer than two people at pharmacies could be bottled at the factories. Consumers could just grab the bottles they want and go.
Stephen Gradijan
Jan 14 2017 at 1:46pm
George,
Nick Bradley’s was simultaneously “begging the question” (a formal logic fallacy) as well as being defamatory and just plain rude.
David Henderson was presumably unhappy at being defamed and chose to respond in kind with a famous riposte that hopefully might make Nick Bradley realize the error of his ways. However I personally don’t expect Mr. Bradley to issue a mea culpa.
https://en.wikipedia.org/wiki/Begging_the_question
Stephen Gradijan
Jan 14 2017 at 1:52pm
P.S. To add to my last post I want to point out that while Nick Bradley pointed out why he thought that David Henderson was incorrect, he didn’t explain why he thought that his disagreement with David Henderson was the result of dishonesty on the part of DH; he simply assumed it.
That unspoken and unexplained assumption is known as “begging the question”.
LD Bottorff
Jan 14 2017 at 1:54pm
George,
When one is accused of being deeply dishonest, it is difficult to reply in a manner that someone won’t consider harsh.
I, for one, did not think that Dr. Henderson was accusing Nick of beating his wife.
Jon Murphy
Jan 14 2017 at 2:09pm
Great article. I think another regulatory reform we could implement is one Alex Tabarrok discusses at Marginal Revolution. Preventing the importation of cheaper drugs, which have already gone through approval processes in other nations, could create significant competition for companies and reduce a huge burden for consumers.
AlanG
Jan 14 2017 at 2:19pm
I will weigh in from the perspective of someone who spent 30 years in the biopharma industry doing regulatory affairs and drug safety. These are just some random comments. FDA approval provides a shield against product liability, witness Merck’s successful litigation strategy for rofecoxib. This also highlights a key fact about the drug approval process in that a complete safety profile of any drug is never known. FDA makes safety decisions based on available data and requires companies to submit ongoing safety information for the life of the drug. Most drug labels are updated regularly with new safety information so removing the FDA from this is dangerous.
@scott sumner – advertising spending is shifting from the MD to the consumer as you see in the large number of print and television ads. “Ask your physician whether drug X is right for you” is the new mantra. do you think the average citizen can make a decision about any of this, particularly when the litany of side effects is gone over?
The assumption that most MDs are fully versed with respect to Rx drugs is a big stretch. Most don’t even look at the drug label and have little idea of the adverse effect and benefit profile. Pharmacology courses in medical school are quite superficial in this regard.
Off label prescribing mainly takes place with older off patent drugs where there is no sponsor who would want to take the time and submit a dossier to FDA as there is no financial incentive.
Saying that almost everyone would not want to take a drug that has not been proven effective is an astounding statement. If this only refers to drugs where there is a documented history of use maybe I can buy it. But what evidence is there? I well remember the case of laetrile and its use as an oncology agents. It never worked. Just because there is a journal article or blogs doesn’t mean a drug works.
The Tabarrok article on pediatric off label use is highly misleading. I was chair of a multi-disciplinary work group on this topic about 15 years ago. There are many reasons why this is the case that would take a fair amount of blog space. I’m happy to cover this in a 2nd post if anyone is interested.
Nick Bradley
Jan 14 2017 at 2:20pm
It’s an astounding GS&A overhead; it’s paying former beauty queens to get doctors to prescribe OxyContin.
Jim Glass
Jan 14 2017 at 2:38pm
As it happens I just came from an interesting discussion of this, “How drug prices are actually set“, started off by a person involved in setting prices for pharma companies and who gives the insider’s view, whose comments were pretty interesting.
Apart from that, really the economic short story behind high pharma prices is that the drug companies use patent monopolies to price discriminate, charge the highest price possible for every customer. So the public list price here will be toppest possible dollar, but all kinds of hidden and disguised lower prices will be given to group purchasers, etc., lower yet to hardship cases, with different ranges of prices in different countries abroad.
Price discrimination like this offends many, but is near universal, everybody does it — nobody does it as well as colleges and airlines. And it increases social welfare when it increases sales (production) as is explained by Tyler and Alex specifically in the case of pharma (and colleges) in this nice video over at MR University.
Now, government could revoke the patent monopoly rules or otherwise force the sale price of drugs down to marginal production cost, say at the price of M&Ms, as so many demand to reduce the price of drugs to the sick and needy and leave so much more money in the pockets of us all. With marginal production costs covered the drug companies would still survive and be profitable.
But … then the issue arises of how to finance the future production of new drugs at about $2.6 billion per successful pop. Eli Lilly reportedly just lost a cool $1 billion on the failure of its Alzheimer’s drug Solanezumab, and has so far spent $3 billion on Alzheimer’s research with no product at all to show for it. There have been over 100 failed Alzheimer’s drug trials and zero successes. But we all want an effective Alzheimer’s drug waiting for us when we need it, don’t we? How will its creation get financed if we decide to help ourselves to today’s drugs at the price of M&Ms?
Of course there are alternatives, e.g., taxpayers could finance the $2.6 billion per drug directly to the drug companies via research subsidies financed on April 15th — but that’s just moving the cost from one pocket to the other. There’s no free lunch here.
That’s reality, from here is my personal subjective opinion…
As to the FDA, in light of the above, my main concern is that it is institutionally biased to delay and obstruct the use of effective drugs and harms and kills people that way, not that it raises prices so much. First, price is set by supply-demand, not cost of production. As long as drug companies can be monopolists engaged in (socially beneficial) price discrimination, prices for effective drugs are going to be very high. Reducing the cost of trials will only increase their profit margins (a good thing to the extent reinvested in getting that Alzheimer’s drug). Second, simplifying trials etc does zip nothing to actually ease creating the drug in the trial. It’s not like if the FDA eased drug introductions then 15 of those 100 tested Alzheimer’s drugs would have worked. Lilly would still be out billions.
I’m all for the FDA easing introductions and use restrictions on drugs, strongly. But I don’t see how it would have any major effect on the price of drugs, a second-order effect, maybe. As to the cost of drug development and pricing of drugs, there’s not much of a free lunch at the FDA either. One person’s opinion.
Jim Glass
Jan 14 2017 at 2:56pm
Nick Bradley writes:
It’s an astounding GS&A overhead; it’s paying former beauty queens to get doctors to prescribe OxyContin.
Yo, Nick:
Putting girls in bikinis in advertisements for cars, beer and GoDaddy web hosting may have nothing to do with the quality of the product, but is *not* overhead.
To the contrary, it spreads the cost of real overhead over the larger number of sales it produces, and thus enables prices to be reduced.
Learn a little bit about business. Hey, I explained this for you in a really simple example earlier. Try to keep up.
BC
Jan 14 2017 at 2:58pm
Everytime someone tries to rationalize the FDA efficacy prohibitions, I do the following thought experiment. Suppose a doctor wanted to prescribe a new cancer drug that the FDA hadn’t yet found effective, only safe. Now, suppose that white cancer patients were allowed to take the drug, but black patients weren’t. I think almost everyone would find this law highly discriminatory against blacks. It would be unthinkable that someone could be denied treatment based on race. No one would say that the law actually would discriminate against whites because they wouldn’t be “protected” from ineffective drugs like blacks would be.
This thought experiment reveals that the efficacy rules aren’t actually “protecting” patients. Patients can already choose to not take the drug and doctors can already choose not to prescribe it. Prohibiting the drugs for everyone effectively puts everyone on the disfavored, not favored, side of discrimination.
AlanG
Jan 14 2017 at 3:23pm
Jim Glass – go visit Dean Baker’s site at CEPR. he has a decent proposal for ending patent monopolies on drugs and funding research such that there would still be new drugs. I don’t happen to agree with him and we have had some interesting discussions both on his blog and off of it.
I have lots of friends both in and out of the pharma industry on the research and drug safety side of things and they all agree that setting market prices is opaque. Gilead just made a guesstimate of what the market would pay for the first Hepatitis C drug ~$80K for the course of treatment and it stuck.
You are correct about Alzheimer’s research. I don’t see much progress on that front until we really know about the biology of the disease and can develop a good surrogate endpoint for clinical trials. Relying on cognition and the loss of mental capacity is subjective and always open to interpretation (there is a consortium trying to develop neural imaging techniques that might aid in trial design). It also leads to ineffective drugs and longer trials to show efficacy. Now some might say that if the drug is shown to be safe why not market it? With a huge patient population of elderly that is growing, chances are very good that there will be huge amount of money paid out of the healthcare system for a drug that doesn’t work. Careful design of clinical trials and FDA approval far outweigh the economic loss IMO.
A good drug development plan coupled with good data lead to speedy FDA approvals. Unfortunately, not every company has a good plan, nor is the data good enough. I wouldn’t rely on simple journal publication of trial results as there is a lot of junk that gets published that consists of under powered trial data.
One final point on drug pricing. the retail price is just that and it is seldom paid. Insurance plans, the VA, Medicaid all get discounts and rebates that add up to a significant amount of money.
David R. Henderson
Jan 14 2017 at 4:51pm
@Jim Glass,
Thank you for your thoughtful comments.
@George,
That seems like an overly harsh comment. You are usually polite in your discourse. Did I miss something?
I don’t know if you missed something. When someone accuses me of dishonesty, and I know it’s not justified, I get harsh. I do disagree with you about whether it’s overly harsh.
Also, once the person makes such a nasty false accusation, I do not converse with that person further.
@Alan G
FDA approval provides a shield against product liability, witness Merck’s successful litigation strategy for rofecoxib.
That’s a good point. And, under Charley’s and my proposal, drug companies would still be free to seek FDA approval.
Saying that almost everyone would not want to take a drug that has not been proven effective is an astounding statement.
I don’t think you meant to put that first “not” in there. On that assumption, I’ll respond. Our point in the piece is that this happens a lot with drugs: many drugs are used for off-label uses. When my doctor prescribed Tagamet for me in the 1980s, for example, it was for an off-label use. It had been approved only for short-term use, not long-term use. There are many such instances. Ask yourself this: have you ever taken a drug for an off-label use?
AlanG
Jan 14 2017 at 5:48pm
@David Henderson,
Yes, good catch the ‘not’ was not supposed to be there. I think from a product liability view, all major pharma companies would continue to seek FDA approval.
Yes, lots of stuff gets used off label but in some cases this is limiting such as with new targeted oncology drugs that won’t be used for lots of different indications as they would not work. Companies are quick to get the first approval and then add further indications to the drug label as that is “sometimes” key to getting reimbursed. The major case of off label use is with older off patent drugs as there is no incentive to do so. It can also blow back against a company as well. When Pfizer got the approval for the antibiotic, trovafloxacin, I think they set a record for the most approved indications for a first approval. Unfortunately, the drug had really bad liver toxicity that was apparent only after post-marketing data began to come in. It’s still on the market but only for some narrow uses these days.
Yes, I’ve taken one or two drugs off label but I consider myself an super-educated consumer and I always read the full prescribing information of any new drug that I am considering. For those interested, all drug labels are on a website run by the National Library of Medicine called ‘DailyMed’ (don’t know if I can post links but a Google search will get you there quickly).
I’m really sympathetic to the point you are raising and most of my working career was spent on ways to streamline drug development, the regulatory process and the understanding of adverse drug reactions (which are far more common than most realize)>
Stuart Buck
Jan 14 2017 at 6:42pm
“Most of this cost is due to FDA regulation.”
Another way of putting it is that most of the cost is due to the need to produce evidence that drugs actually work, rather than being useless or even outright harmful. We could, in theory, go back to the era of snake oil (which is what we still have w/r/t herbal supplements), but there are good reasons (rampantly asymmetrical information, for example) that we abandoned that approach several decades ago.
“Some potentially helpful drugs don’t ever make it to market because the cost the company must bear is too high.”
This is an argument for finding cheaper ways to do clinical trials.
“If we simply went back to pre-1962 law, the FDA could still require proof of safety, but would not be able to require evidence on efficacy.”
This is a common suggestion, but it fails for the basic reason that no drug is “safe.” There is simply no such thing. All drugs have side effects, and some are particularly dangerous. Cancer drugs are the best example. Chemotherapy was originally developed from mustard gas used in World War I, and some of the “best” chemo drugs we have today are incredibly toxic (google “adriamycin” and “heart failure”). The ONLY reason we would ever give a chemo drug to someone is NOT that it’s safe, but that there is rigorous evidence that there might be a benefit large enough to outweigh the toxic and sometimes fatal side effects. Saying that we should approve chemo drugs for safety without efficacy is impossible nonsense.
“This one change would allow drugs to be developed faster–often as much as 10 years faster. Market success would establish efficacy.”
Is market success the same as a randomized experiment showing efficacy? Economists have one some randomized experiments showing that charter schools in urban areas are effective. Were those economists wasting their time looking for anything but market efficacy?
Alex
Jan 14 2017 at 7:10pm
Great Article.
Notice how your critic Nick doesnt dispute the over $2 B cost to bring a drug to the market. He just talks about marketing costs, something totally unrelated.
And Stuart, two things:
“This is an argument for finding cheaper ways to do clinical trials”
Such as? Until you show us a way of doing this this criticism is invalid.
Also: “The ONLY reason we would ever give a chemo drug to someone is NOT that it’s safe, but that there is rigorous evidence that there might be a benefit large enough to outweigh the toxic and sometimes fatal side effects”
Exactly, and why should the FDA make that call and not the patient? When David talks about safety, he doesn’t mean that the drugs have no side effects, but that the side effects are clear so the patient knows them and knows the risk. For someone who is dying fast, side effects are not as relevant as to someone who is healthy.
Michael Byrnes
Jan 14 2017 at 11:55pm
Stuart Buck wrote:
This is actually an area where the much-maligned 3rd party payment system is actually a plus.
Eliminating the requirement for FDA approval would absolutely not mean a return to the era of snake oil, at least not for the expensive drugs that most people cannot afford without insurance.
Health insurers don’t simply pay whatever cost the pharma company demands for its cool new drug – nowadays payers are more sophisticated than that, and the whole field is moving towards value-based pricing systems.
A lack of evidence that a drug works is going to limit what the pharma company can charge for it.
Thus, FDA or not, there will always be efficacy trials.
The potential for abuse, I think, would be for less costly drugs – ones that a lot of people could afford out-of-pocket. There is some potential for snake oil there.
One change that I think would be interesting is this: how about requiring FDA approval before a pharma company is allowed to engage marketing/advertising to patients? Let them talk to physicians, health systems, insurers without FDA approval, but if they want to run those “Ask your doctor about our new drug…” ads they still need FDA approval.
Peter
Jan 15 2017 at 2:36am
Interesting discussion here.
Can somebody please explain to me or point me to some literature that explains in detail how doing away with the proof-of-efficacy requirement would reduce drug developing costs appreciably? I have heard this assertion many times but I have never seen an explanation of it. The trials that are reliable enough for assessing safety are not good enough for assessing efficacy? Why not?
Also, what Stuart wrote above, namely, that a drug’s safety profile is usually judged in the context of its efficacy (because there are almost always undesirable side effects) seems to go against the argument that one can sensibly judge the safety of a drug without considering its efficacy.
David H. & Charles Hooper wrote:
“If we simply went back to pre-1962 law, the FDA could still require proof of safety, but would not be able to require evidence on efficacy.”
How was proof of safety before 1962 provided? If by randomized controlled trials, why could the same trials not be used to assess efficacy as well?
Nathan Benedict
Jan 15 2017 at 9:10am
Peter–I’m no expert in this field, but it seems obvious to me that the work required to prove that a drug is safe is far less than that required to prove that it is efficacious. A clinical trial for safety measures deaths and side effects, presumably compared to a placebo (and can often be done on healthy people). A clinical trial for efficacy measures improvements in a whole host of factors, usually compared to an existing drug that we know works to some extent (and requires participants suffering from the disease sought to be cured).
To draw an admittedly imperfect analogy, consider making a movie. How many Hollywood movies do you see that contain glaring technical errors like boom mikes in the shot, or actors flubbing their lines? Almost none. How many movies do you see that just plain stink? Lots. Proving safety is like lining up the scene so the mike isn’t visible. Proving efficacy is like writing a good story, with good actors, good directors, good editing, good special effects, etc. The difference is night and day.
Stuart Buck
Jan 15 2017 at 9:54am
Alex —
On cheaper clinical trials, look up the TASTE trial if you want to see how it’s done.
And this makes no sense: “Exactly, and why should the FDA make that call and not the patient? When David talks about safety, he doesn’t mean that the drugs have no side effects.”
Then what does it mean to approve for safety? What exactly does that look like, if you’re the FDA commissioners? If adriamycin/doxorubicin were up for approval solely on safety grounds (without evidence of efficacy), all you would see is a drug that can cause severe nausea, alopecia, hoarseness, joint pain, difficult urination, shortness of breath, stomach pain, and sometimes heart failure leading to death.
Nothing good about any of that. Now, on what grounds do you approve that drug? Remember, efficacy is out of bounds, you’re not allowed to demand clinical trials that show efficacy. All you can see is the safety data.
AlanG
Jan 15 2017 at 10:43am
Folks need to know that drug safety is ongoing and most drugs do not have a complete safety profile upon approval. Look at the example of the Cox-2 anti-inflammatory drugs that were thought to be much safer than the older class of drugs. Several years after approval cardiac events were noticed and two of the drugs were pulled off the market. Pfizer funded a very large safety study for celecoxib that was just completed this year (two years after the drug went off patent) that showed the drug was not any less safe than ibuprofen or naproxen. Companies spend significant amounts of money on post-market surveillance of safety and many changes to the drug label take place after approval to update safety issues.
If the intent is to allow drugs with unproven efficacy to be sold should informed consent on the part of the patient be required? This is what is required of experimental drugs during clinical trials. IMO, more use should be made of FDA’s Treatment IND regulations that allow companies to recoup costs while a drug is still in clinical development. Access can be expanded but efficacy is still studied. Companies don’t like this regulation as they have to provide FDA with pricing information and cost to assure that no profits are being made.
Several commenters ask about studying safety and efficacy at the same time. Of course this is what happens. the only pure safety trial is Phase 1 where enough information is compiled to demonstrate that the drug can be used in clinical trials with a knowledge of some of the major toxicity issues.
Thereld
Jan 15 2017 at 11:16am
Thank you, this is a good introduction to a topic with which I’m unfamiliar.
Just one question about:
Can we get a source on that?
Stuart Buck
Jan 15 2017 at 12:02pm
Michael Byrnes may be correct that we could move to a world where instead of the FDA, insurance companies all demand the same level of proof (via RCTs) before they pay for a drug/device/treatment. That said, even RCTs are very easy to manipulate in dozens of different ways (choice of comparator, dosages, blinding, subgroup analyses, even just cleaning data for analysis). Insurance companies do not have the internal expertise to make sure that RCTs are performed and analyzed in a rigorous way, nor would it be efficient for them all to do so one by one. So we could imagine creating a centralized organization that would monitor how RCTs are done, and would take a second look at the data. In fact, this would be what the FDA does right now.
Put all of this together with Jim Glassman’s excellent point: “Second, simplifying trials etc does zip nothing to actually ease creating the drug in the trial. It’s not like if the FDA eased drug introductions then 15 of those 100 tested Alzheimer’s drugs would have worked. Lilly would still be out billions.”
Exactly. Even if we got rid of the FDA entirely, and moved to privatize the generation of evidence, drugs would still be expensive to develop, for the simple reason that human biology is hard, and drugs that look promising in the early stages (such as preclinical work) often or usually fail to do anything for humans.
Mark Bahner
Jan 15 2017 at 1:28pm
I don’t see how this explains the chart that David Henderson presented…an increase from less than $200 million in the 1970s to almost $2.6 billion in the 2000s.
Presumably, we’re learning more and more about the human body, and more and more about how drugs interact with the human body.
With most things in the world (e.g., solar cells, wind power, oil fracking, OLED TVs, etc.) more knowledge means *lower* costs.
Dylan
Jan 15 2017 at 1:48pm
@ Stuart
Exactly. The reason that drug prices are high is because we don’t really know how to develop drugs and make sure they work in an efficient way. Until we do that everything else is going to be fiddling around the margins at best, or a giant trade-off between cost and evidence based medicine. There’s some progress being made with things like adaptive trials and personalized medicine approaches that would allow for smaller trials and smaller patient populations, but those are still early as far as I know.
@BC
They actually do something like this already. Lots of drugs will have restrictions on the patient population, which sometimes includes race. And sometimes it isn’t there, but maybe should be. For instance, there are reasons to believe that Plavix is not very effective among patients with a certain genetic marker that is pretty common among Asians, but doctors don’t test for this marker before prescribing and just give to everyone equally and try to catch problems by monitoring them closely afterwards.
@ Dave Henderson
It’s probably worth at least acknowledging that the $2.6B figure is on the high side as far as these estimates go, and that number includes the direct costs, the cost of failures (9 out of 10 drugs fail and I chalk this up more to the fact that we don’t really know what we’re doing a lot more than I think it’s the FDA getting in the way), and also the WACC. All of those need to be included to get an accurate model for sure, and the estimates that exclude these costs are going to be much further off, but still WACC is somewhat subjective and given that it is the largest component of the $2.6B I think it’s still worth treating that number with some skepticism.
David R. Henderson
Jan 15 2017 at 2:40pm
@Dylan,
It’s probably worth at least acknowledging that the $2.6B figure is on the high side as far as these estimates go
It’s definitely worth acknowledging if ti’s true. What other estimates do you have in mind? Cites please.
Stuart Buck
Jan 15 2017 at 3:19pm
Cite: https://www.nytimes.com/2014/11/19/upshot/calculating-the-real-costs-of-developing-a-new-drug.html?_r=0
Stuart Buck
Jan 15 2017 at 3:30pm
Mark — “Presumably, we’re learning more and more about the human body, and more and more about how drugs interact with the human body.”
One answer is that we’ve already found most of the low-hanging fruit. We know more and more, but that doesn’t mean that every disease actually has a druggable target, etc., etc., etc. See https://www.scientificamerican.com/article/outsmarting-cancer/ for example.
Dylan
Jan 15 2017 at 4:26pm
@ David Henderson
The below link is for a DNDI estimate that puts their estimate at 100-150m EUR for a NME including the cost of failure.
http://www.dndi.org/2013/media-centre/press-releases/dndi-rd-model/
I am not presenting this link suggesting that it is the right number, indeed I think it is almost certainly farther from the true average than the Tufts study due to methodological issues that are too numerous to issue here. But it does present a pretty accurate lower bound for a certain type of drug discovery, and the Tufts study is an upper bound for the most expensive type of drug discovery we know how to do, product developed start to finish inside one of the largest global pharmaceutical companies in the world. There’s a reason after all that the global pharma have slashed their internal R&D, they found it cheaper to acquire drugs than to do it themselves.
Stuart linked to the NYT piece that gives a good overview of some of the issues with the Tufts study. Here’s some similar points raised from more of an industry insider, who’s largely sympathetic to the Tufts study.
https://lifescivc.com/2014/11/a-billion-here-a-billion-there-the-cost-of-making-a-drug-revisited/
ColoComment
Jan 15 2017 at 5:16pm
Some thoughts:
1) drug patents START when a drug is invented, not when it’s approved by FDA. Which can/does result in an abbreviated monopolistic time within which a drug manufacturer can sell the product and recoup costs before the generics come on line. One solution might be to issue a provisional type of patent when application is made to FDA, but extend the full patent lifespan to x years from and after FDA approval.
See here (this is from 2002, so if the patent situation has changed, please correct):
http://www.forbes.com/2002/05/02/0502patents.html
2) Someone above mentioned the (a) low-hanging fruit, and (b) the ~90% failure rate.
I have a vague recollection of reading that drug manufacturers are now foraging far and wide to find the “higher fruit,” in jungles and tundras, oceans and alpine, for plant & animal products that may lead to production and commercialization of human drugs. With respect to drug failures, we need to recall that the cost for a drug to fail either safety or efficacy trials is the same as for those that succeed. But the only benefit to the manufacturer is that it’s identified a dead-end. Dead-ends don’t make payroll or keep the heat on in the labs.
3) Re: marketing. I don’t know when drug manufacturers began marketing directly to consumers, but they must perceive some benefit or they wouldn’t continue to do it. The biggest problem with that is captured in a statement attributed to John Wanamaker, founder of the department stores: “Half the money I spend on advertising is wasted; the trouble is I don’t know which half.” (John Wanamaker, (attributed) US department store merchant (1838 – 1922)
4) The EpiPen situation did nothing to assuage the public’s negative view of drug manufacturers as greedy ba$tard$ whose goal in life is to squeeze every penny possible out of consumers who must buy their product or die. (hyperbole used for emphasis)
Peter
Jan 15 2017 at 5:29pm
David and Charles write “Most of this cost [the cost to get one drug to market successfully] is due to FDA regulation.”
According to the graphic at the beginning of the blogpost, drug development costs have increased tremendously over time. For instance, they were 6 times higher in the first decade of the 21st century than in 1980 (taking the data in the graphic at face value). For FDA regulation to mainly be responsible for high drug prices, FDA regulation must also mainly be responsible for the dramatic increase in the cost of bringing a drug to market? How so? Has the modus operandi of the FDA changed enough to explain the increase? Did FDA approval standards become more demanding? Did drug approval times increase (I think the opposite is the case)?
Alternatively:
1. The data are not correct.
2. The low-hanging fruits have been picked and it has just become more difficult to find drugs that work (irrespective of the FDA). See for instance here:
http://marginalrevolution.com/marginalrevolution/2016/12/depressing-paper-great-stagnation.html
Jim Glass
Jan 15 2017 at 7:02pm
Regarding challenges to the Tufts $2.6 billion number, two thoughts: (1) The Times’ criticism of it is rather odd (besides being outdated, the full study has been released); and (2) the exact number — whether “Pharma’s own” $2.6 billion or Nader’s “mere” $161 million — really doesn’t matter (except in political PR wars).
(1) The Times writes …
OK, if it’s pefectly sound, what’s the problem with it?
Well, yeah. If investors in the pharma companies see they aren’t going to recover the their $1.2b (or whatever) in opportunity cost per new drug, and thus sell out to invest in other things, that’s going to be the end of these companies developing new drugs. That’s the point!
Maybe those to whom this idea “rings false” need an education as to the truth of it – instead of having their strange belief sympathetically repeated by a Times writer who in the very previous paragraph knew better.
(2) More fundamentally, high-ball and low-ball numbers thrown around for the cost of drug development are just PR devices in the political fight over whether or not pharma firms are to be deemed rapacious profiteers reaping big profits by exploiting the sick and weak, and have their incomes (prices) slashed by law accordingly.
Yet whether they are or not can be coolly checked by examining industry financials.
E.g. NYU/Stern (my alma mater) reports that the Pharma sector of the stock market has a forward Price Earnings ratio (stock price/expected future earnings) of 21.19, versus the Total Market’s 46.11. There’s hardly any evidence of any exceptional profiteering in that, whatever the cost of developing new drugs may be.
For comparison, other sectors with higher forward PEs include: Soft drinks 52.36, cable TV 27.17, education 36.03, environmental waste services 28.63, machinery 26.46, and trucking 21.82.
So, gut the return on drug development for political purposes, and the investors who fund it may decide to move on instead to a diversified portfolio of funding trucking, environmental waste services, and developing new soft drinks, and do much better for themselves.
Stuart Buck
Jan 15 2017 at 8:03pm
Two facts that are relevant to this debate:
1. Gleevec is a great drug for chronic myeloid leukemia. When it first came out, the FDA was so eager to get it to market that the approval came in a mere 2.5 months, after a few Phase II trials that didn’t even have a control group! Moral: when a drug actually works, the FDA is the least of anyone’s concerns.
2. By contrast, the average solid cancer drug approved between 2002 and 2014 led to a survival benefit of a mere 2.1 months. http://jamanetwork.com/journals/jamaotolaryngology/fullarticle/1891387 To make matters worse, these clinical trials overstate the real-world benefits: https://www.statnews.com/2016/11/21/cancer-clinical-trials/
Given these very slight benefits, it is impossible to say that the FDA is setting the bar too high. To the contrary, FDA could hardly set the bar any lower before it starts approving drugs that literally have no benefit.
It is interesting that one side of this debate seems to be unfamiliar with how the FDA actually works or what it has been approving, let alone how medicine works either. Indeed, one might think that some folks have never even met or heard of a cancer patient. My wife had breast cancer several years ago, at age 34. When the nurses were giving her an IV of adriamycin, I was astonished to see that the bag of red fluid was covered with labels warning that it was a “biohazard,” and of all the dire consequences that would happen if it came in contact with a human person (tissue death, etc.).
If you know anything about actual cancer drugs and how toxic they are, the notion of approval based on “safety” seems delusional.
AlanG
Jan 15 2017 at 8:57pm
I worked with Joe DiMasi of Tufts on a couple of projects while I was still active and he is pretty up front about all the assumptions that go into his cost of development model. He’s published a paper or two that go into things in more detail. He is working with financials that are provided directly to him from pharma companies so his work is based on that information. Obviously, one can use different modeling and come up with other valuations. the reference that Dylan provides above is a useful analysis of the Tufts model.
Addressing Peter’s point about changes in FDA regulation since 1980, the answer is sometimes, particularly in the safety area. We know a lot more about mechanisms of toxicity these days and certain classes of drugs will necessitate a longer clinical trial in order to monitor for safety. The efficacy standard has not changed at all and in fact the FDA is willing to use more modern endpoints and methods of analysis. IMO, there is far more certainty now regarding trial design for efficacy with most drugs than was the case in 1980. FDA continues to adopt new science once it is validated.
Stuart Buck’s point about needing a central organization to review data is right on the mark. Regulatory filings for a new drug are huge and contain the full data set that addresses both safety and efficacy. I recall during one of the negotiations with FDA over reauthorization of the user fee program analyzing the man hours and cost to FDA to review a New Drug Application. The data I had was from 2003 and the cost to review an NDA was $2.1M which at the time was about 12 full time employees working a full year. Of course the work load is spread out over many employees and the review times have gone down considerably over the years.
Alex
Jan 15 2017 at 8:58pm
“Now, on what grounds do you approve that drug? Remember, efficacy is out of bounds, you’re not allowed to demand clinical trials that show efficacy. All you can see is the safety data.”
You would approve it nevertheless. In fact, if it were for me, I would end the war on drugs and let any adult buy any drug he wants. Frankly, I see no role for the FDA at all.
Think of this case. A drug has a 50% chance of saving your life and 50% chance of killing you. You are sick, dying fast. You are willing to take the chance. Do you think an FDA bureaucrat would approve this drug? No, he would not, and your 50% chance of surviving becomes 0% chance.
Stuart Buck
Jan 15 2017 at 9:38pm
“You would approve it nevertheless. ”
You’re just arguing for no FDA. But you’re not defending the (nonsensical and ignorant) position that the FDA could approve based on safety.
Stuart Buck
Jan 15 2017 at 11:29pm
By the way:
“I would end the war on drugs and let any adult buy any drug he wants. ”
It is important to clarify that the FDA is not the same as the DEA. Doctors are free to prescribe whatever they want, and patients are free to ingest whatever they want. What the FDA does is tell drug manufacturers when they are and are not allowed to *market* a drug as having been proven to remedy a particular condition. The FDA is more like an anti-fraud-in-marketing agency than anything else.
Peter
Jan 16 2017 at 2:19am
I’m surprised to see Stuart write that:
“What the FDA does is tell drug manufacturers when they are and are not allowed to *market* a drug as having been proven to remedy a particular condition. The FDA is more like an anti-fraud-in-marketing agency than anything else.”
The FDA does not just regulate how any given drug can be marketed. No, only drugs that are approved by the FDA can be marketed in any shape or form. Hence, doctors can prescribe whatever they want from those drugs that the FDA let on the market.
As to the last post by Jim Glass:
1. Yeah, hearing economically illiterate tirades against pharma companies is tiresome.
2. Though pharma profits are not low compared to other industries. Princeton University health care economist Uwe Reinhardt wrote in 2001:
“relative to those of other industries,the pharmaceutical industry’s profits may be on the high side, but in the absolute they are not large enough to offer much relief for any cost containment effort, as they constitute only a minute fraction of total national health spending.”
Perspectives On The Pharmaceutical Industry. Health Affairs, 2001
http://content.healthaffairs.org/content/20/5/136.full.pdf
I suspect that assessment is still true.
Maybe of interest as well:
Pharmaceutical industry gets high on fat profits. BBC News , Nov 2014
http://www.bbc.com/news/business-28212223
Stuart Buck
Jan 16 2017 at 9:37am
“Hence, doctors can prescribe whatever they want from those drugs that the FDA let on the market.”
Mostly true. But sometimes doctors will prescribe medications (such as domperidone) that are not FDA-approved, and the patient then gets the drug from international pharmacies or from a compounding pharmacy in the US. Other doctors then debate as to whether it’s a good idea to do so (see http://thehealthcareblog.com/blog/2013/08/26/should-a-doctor-prescribe-drugs-that-are-unapproved-by-the-fda/).
Nathan Benedict
Jan 16 2017 at 9:49am
Stuart Buck wrote: “It is important to clarify that the FDA is not the same as the DEA. Doctors are free to prescribe whatever they want, and patients are free to ingest whatever they want. What the FDA does is tell drug manufacturers when they are and are not allowed to *market* a drug as having been proven to remedy a particular condition. The FDA is more like an anti-fraud-in-marketing agency than anything else.”
This could not be more false. Please read https://en.m.wikipedia.org/wiki/Abigail_Alliance_for_Better_Access_to_Developmental_Drugs_v._von_Eschenbach
In that case, the patient wanted to try a particular drug, the doctor was willing to prescribe it, and the FDA refused to allow a voluntary transaction to take place.
[Full URL substituted for shortened URL. Please use full URLs in EconLog comments. –Econlib Ed.]
Hazel Meade
Jan 16 2017 at 10:35am
Nobody has mentioned “reciprocity” in drug approvals between the EU, Canada, and the US.
I recall last year there was some discussion of this. Essentially, any drug approved in the EU would be allowed to be sold in the US, and vice versa. This would significantly increase the number of drugs on the market and lower the relative cost of bringing a drug to market because the effective market would be larger. A drug company would not have to go through separate approvals processes for each region. The Eu also has high standards for approval so this shouldn’t result in a free for all.
There might be some efforts to always apply for approval in the easiest location, but given the backlogs that would cause, I imagine paying a bit extra for faster approval might be worth the cost to many drug companies.
In any case, the EU and US could work out some sort of reconciliation board to synchronize approval standards and adjudicate disputes.
AlanG
Jan 16 2017 at 10:48am
@Hazel Meade – the idea of reciprocity of approvals has been around for over 20 years. My former employer was advocating for this back in 1992. While the idea was interesting back then, it would not have much of an impact as regulatory filings are usually done simultaneously and companies want a US approval as quickly as possible because of the lack of price controls here relative to other countries (though this may change because of Trump’s announcement that he wants to negotiate pricing for Medicare and Medicaid).
Companies already pay user fees in both the US and EU for regulatory review and it has been the case in recent years that 80% of the first approvals are here in the US. The regulatory submissions have been harmonized and there is very little difference in what gets filed (other than in Japan which has some strange requirements).
This is basically an idea that was interesting once but no longer needed. You also run the risk to having insurance companies and other payers not accepting the foreign review which might happen.
Dylan
Jan 16 2017 at 10:56am
@ Alex
“Think of this case. A drug has a 50% chance of saving your life and 50% chance of killing you. You are sick, dying fast. You are willing to take the chance. Do you think an FDA bureaucrat would approve this drug? No, he would not, and your 50% chance of surviving becomes 0% chance.”
In your example it is unlikely we would know the chance the drug has of saving your life, but if we’re going to assume some numbers for the case of the thought experiment I think it is probably more illuminating to say that drug X for cancer has maybe a 5% chance of extending your life by a few months, a 95% chance of giving you severe side effects whether or not it works to extend your life, and maybe a 5% chance of killing you outright. This last part hits close to home as a colleague died last month due to an AE from chemo. It is likely he would have lived at least a couple more years without the chemo.
Effem
Jan 16 2017 at 11:02am
What can we learn from the supplement market? Feels like a wide range of compounds are available at a low cost but that we still have little idea what works and “scams” and fads occur with great frequency. Doesn’t bode well for extreme deregulation from my vantage point.
David R. Henderson
Jan 16 2017 at 11:26am
@Hazel Meade,
Nobody has mentioned “reciprocity” in drug approvals between the EU, Canada, and the US.
Actually, Hazel, Charley and I did in the above-cited study and our proposal is more extensive than the one you make.
Peter
Jan 16 2017 at 12:19pm
Stuart wrote:
“It is interesting that one side of this debate seems to be unfamiliar with how the FDA actually works or what it has been approving, let alone how medicine works either.”
I agree with this, with some qualification.
Take for instance the claim that David Henderson and co-author make (in the linked brief “Why are drug prices so high”) to the effect that if drugs were only approved for safety the market would assess efficacy. They write that “doctors and patients regularly evaluate drugs for efficacy”. That is to some extent true. But it only works for some drugs. Many medical drugs are not like television sets, headphones or a pizza where a consumer can easily assess their quality. Take for instance diabetes 2 drugs (that is, adult-onset diabetes). The drugs that are on the market have all been approved on the basis of their reducing the blood sugar level. However, blood sugar level is just a surrogate marker* that by itself is not important. What really matter are the micro- and macrovascular complications that diabetes can cause (blindness, heart attacks, strokes, etc). And we simply don’t know whether the currently available and widely prescribed expensive patent-protected drugs that reduce blood sugar are any good for reducing the incidence of these micro- and macrovascular complications. Nobody knows this. Why? Because this knowledge is expensive to generate. You would need long-term clinical trials for that (lasting 10 years or so). But the FDA does not require such trials for marketing approval, and hence pharma companies have no interest in doing them because their fiduciary duty is to shareholders and not to sick people. In other words, pharma companies care about making money and not about patient health. If it’s possible they will gladly sell you drug of unknown or zero efficacy (like we also see in the market for supplements). In the case of diabetes 2 drugs both the FDA and the market have failed to establish that the available drugs do any good – not because it is impossible to do so, but because it is expensive. Patients tend to trust doctors, so it is actually cheaper for drug companies to corrupt doctors’ judgment by focusing them on treating unproven surrogate outcomes than on establishing the efficacy of their drugs for patient-relevant outcomes.
Another example is cold medicines**. The available ones don’t work. But they are still sold. The joke is that if you take them your cold will disappear after about 7 days, and if you don’t take them it will take about a week.
Another example is Merck’s drug Vioxx. Did the market establish that for most people it did not make sense to take this drug because the balance of therapeutic effects and undesirable side effects were not as favorable as Merck claimed? No it did not. The evidence of unacceptable side effects of Vioxx (that Merck then could no longer deny) was created by clinical trials that Merck was forced to do by the FDA because Merck wanted to gain approval to market Vioxx for other medical indications. Without these FDA-mandated trials Vioxx would have created even more damage than it actually did. Why did it cause so much damage? Because, unlike David and Charles claim, it’s not always possible or easy for doctors and patients to assess drug efficacy. If patients die from taking a drug you can always blame the patient’s disease. And it cannot easily be established without clinical trials whether the patient was likely killed by the drug or the disease.
Of course, we can deregulate drug approval. But we will still need expensive clinical trials to establish efficacy and side effect profiles of drugs. This knowledge often cannot be generated as an inexpensive side-product of some other valuable process.
* The limitations and potential hazards of using surrogate markers. Feb. 2015
http://www.ti.ubc.ca/2015/02/03/the-limitations-and-potential-hazards-of-using-surrogate-markers/
**Prevention and treatment of the common cold:
making sense of the evidence. CMAJ (Canadian Medical Association Journal), February 18, 2014, 186(3)
Stuart Buck
Jan 16 2017 at 1:36pm
“They write that “doctors and patients regularly evaluate drugs for efficacy”. That is to some extent true. But it only works for some drugs. Many medical drugs are not like television sets, headphones or a pizza where a consumer can easily assess their quality.”
Exactly. Consider the typical cancer drug that extends life by 2.1 months. That is simply NOT an effect that any patient can discern for themselves, and given how much selection bias and just sheer noise there is in real-world data, there is no way that doctors could individually discern such a small effect either.
Yes, for enormous effects (like tobacco > cancer) or immediate and reversible effects (like one’s experience of a migraine), there are ways to learn about a substance’s effects by seeing it before your eyes. But there’s often no substitute for doing actual science, i.e., randomized trials.
Floccina
Jan 17 2017 at 2:43pm
Jim Glass wrote:
If the lower barrier to entry increases the number of products on the market, that should make drugs less expensive. New “me too” drugs that cause fewer side effects, for example can make a drug company reduce the price which is nice for the people not affected the side effect.
DeusExMachina
Jan 17 2017 at 4:08pm
Stephen Gradijan:
If you are going to accuse someone of logically fallacious arguments, you could at least understand basic logic. The answer to which you were referring was most certainly NOT an example of begging the Question.
Review your own citation.
DeusExMachina
Jan 17 2017 at 4:11pm
Jim Glass writes:
Um yes, it is. Just because it leads to greater profits does not make it not overhead. What on earth do you think the word even means?1?
Jim Glass
Jan 19 2017 at 1:03am
What I’m thinking is what I wrote:
“Marketing isn’t a cost driver *at all*. It increases net revenue. Spend $x on marketing, receive $X+ in revenue. As such, it provides funds *for* product development and, if anything, provides leeway to reduce drug prices (through price discrimination).
“Start a business. Don’t market it. Only your mother and three best buddies are willing to buy your product – but not at anywhere near the price you need to cover your startup costs. You are busted. OR instead market it, sell to 10,000 customers at a modest price, reinvest your profits in developing your next products…”
Advertising drugs is *not* an overhead cost — like R&D, salaries, rent, taxes, insurance etc. — that drives *up* the price of drugs, *as charged*. By spreading the fixed costs related to the drug over more sales it *reduces* the lowest profitable price of the drug &/or provides more funds for R&D. Clear enough?
Jim Glass
Jan 19 2017 at 1:24am
Floccina writes that Jim Glass wrote…
“If the lower barrier to entry increases the number of products on the market, that should make drugs less expensive”.
Certainly, but that doesn’t reduce the cost of actually developing the first drug. That’s why I said easing drug introductions might have “a second-order effect” on reducing drug prices.
In some cases it surely has a major effect on price — the FDA’s unexplained blocking of legit competitors to EpiPen being a notable case looked at by Scott Alexander. But overall, in cases where competitors are all on patent and so all able to engage in monopolistic price discrimination, I gotta think the price effects of competition will be limited.
Sorta like the competition between top colleges for students. Kids from well off families never get anything like the marginal cost price.
Jim Glass
Jan 19 2017 at 1:08pm
As to the price of drugs and the rate of drug development, here’s the economically literate medical doctor (psychiatrist) Scott Alexander again…
~ quoting ~
So by my count, there are eight-and-a-half studies concluding that price regulation would hurt new drug innovation, and one-half of a study concluding that it wouldn’t …. One source I do trust is RAND … they write:
…let me put this in context. In 2060 there will probably be 420 million Americans and 523 million Europeans … So about a billion people each losing about 0.7 years of their life equals 700 million life-years. Since some people live in countries outside the US and Europe and they also benefit from First-World-invented medications, let’s round this up to about a billion life-years lost.
What was the worst thing that ever happened? One strong contender is Mao’s Great Leap Forward, in which ineffective agricultural reforms and very effective purges killed 45 million people … [estimated human cost] 45 million * 20 = 900 million life-years…
RAND’s calculations plus my own Fermi estimate suggest that prescription drug price regulation would cost one billion life-years, which would very slightly edge out Communist China for the title of Worst Thing Ever…
Am I exaggerating or being facetious? I’m actually not sure … The only thing I can say in my own defense is that I am acknowledging that the question exists.
Comments are closed.